

Overview
The focus of my current research is on computational models of microbial ecosystems. I am also interested and have recent publications on deep learning, origin of life, epidemiology, genome evolution, and structure and function of biomolecular networks. I am a physicist by training and often rely on statistical physics modeling techniques. I particularly love simple-yet-rich “bottom-down” models.
In my papers I am trying to shed some light on the following questions:
– How microbial ecosystems achieve and maintain their diversity? How many stable states do they have? How to efficiently control complex microbial ecosystems? How phages co-evolve with their microbial hosts?
– How dynamical properties of biomolecular networks affect their functioning inside living cells? How they achieve robustness against noise and perturbations, minimize crosstalk, and limit the effects of non-specific interactions?
– How genomes and networks emerged at the dawn of life and continue to change in the course of evolution? What is the role of Horizontal Gene Transfer at different timescales of bacterial evolution?
My research has practical applications in manipulating the human gut microbiome, controlling regime shifts in oceans and soils, mitigating epidemics of pathogens and infectious diseases such as COVID-19, and analyzing big data generated by genomics and systems biology projects.
Microbial ecosystems
In (Nat Comm 2021 Nov, Nat Comm 2021 Feb, PLoS Comp Bio 2019, ISME 2018, eLife 2019, PRL 2018) we computationally modeled microbial communities with species competing for nutrients and/or cross-feeding metabolic byproducts to each other. This cross-feeding is particularly important in the human gut microbiome – a quintessential example of the complex microbial ecosystem. Its ~600 microbial species identified in different individuals are known to exchange with each other ~250 metabolites (Nat Comm 2021 Feb, PLoS Comp Bio 2019, ISME 2018).
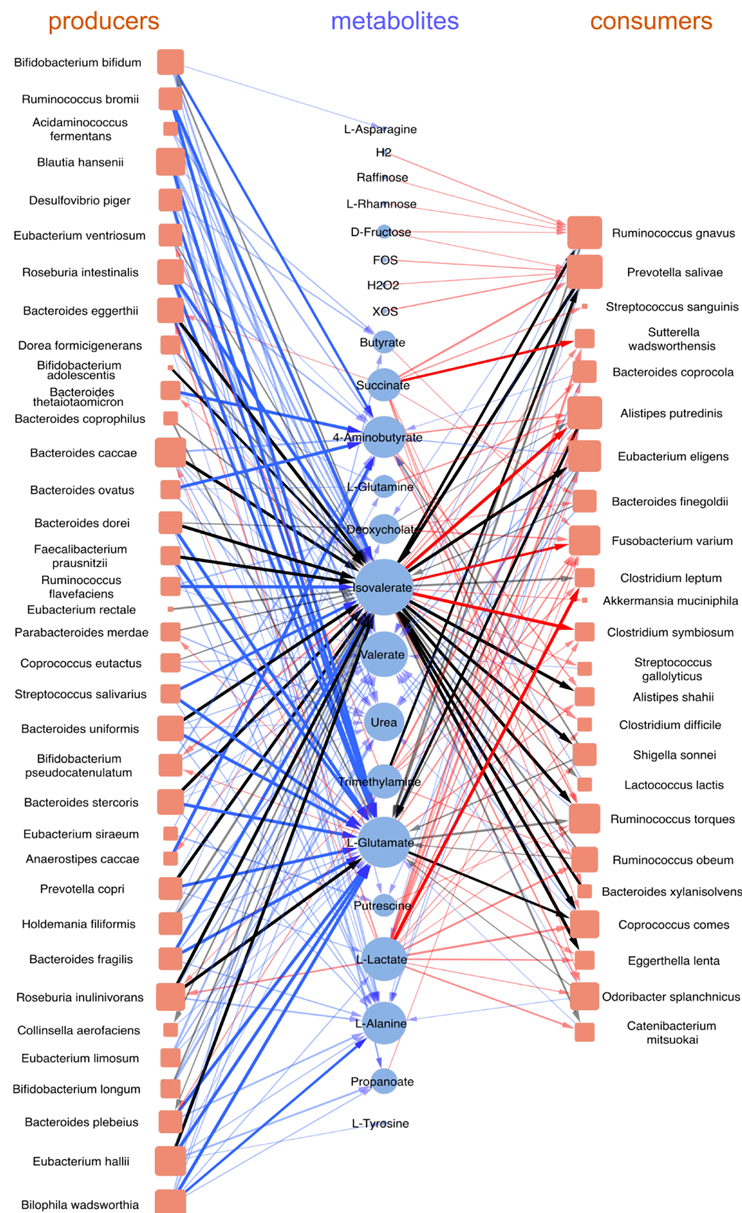
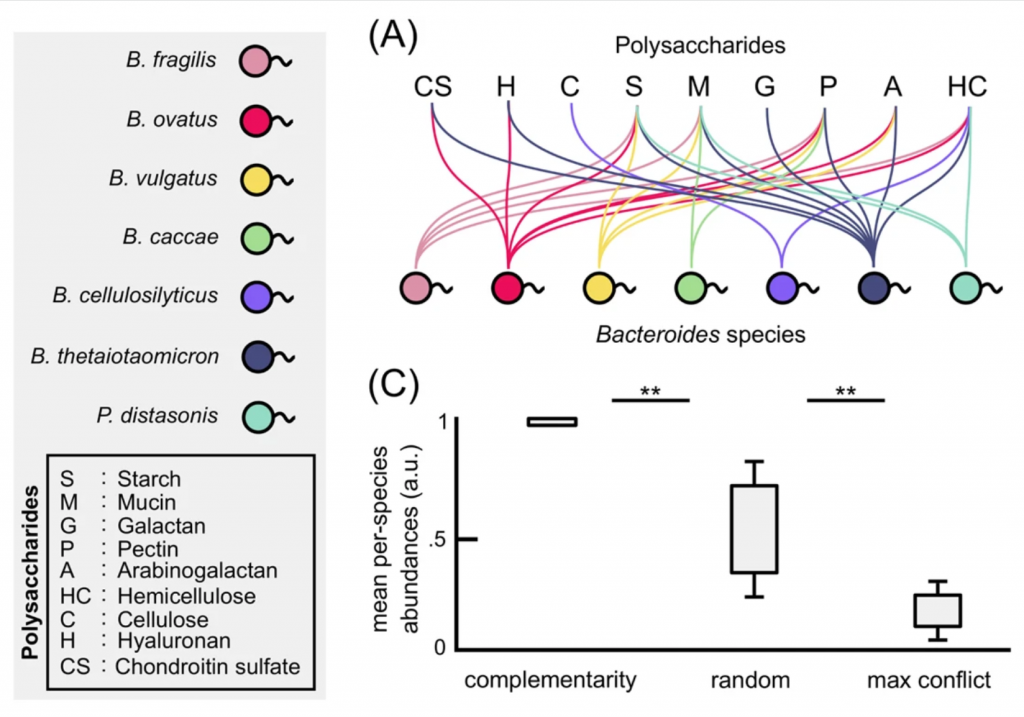
We applied the stable marriage/matching problem (2012 Nobel Prize in Economic Science) to understand multistability and regime shifts in several types of microbial communities (ISME 2018, eLife 2019, mSystems 2019).
- In the first type of a community (Nat Comm 2021 Nov, ISME 2018) resources are utilized sequentially (diauxie) in the order determined by each species regulatory network. The population dynamics of such communities realized in serial dilution experiments, as well as in naturally occurring boom-and-bust cycles (e.g., in the upper ocean microbiome of temperate regions).
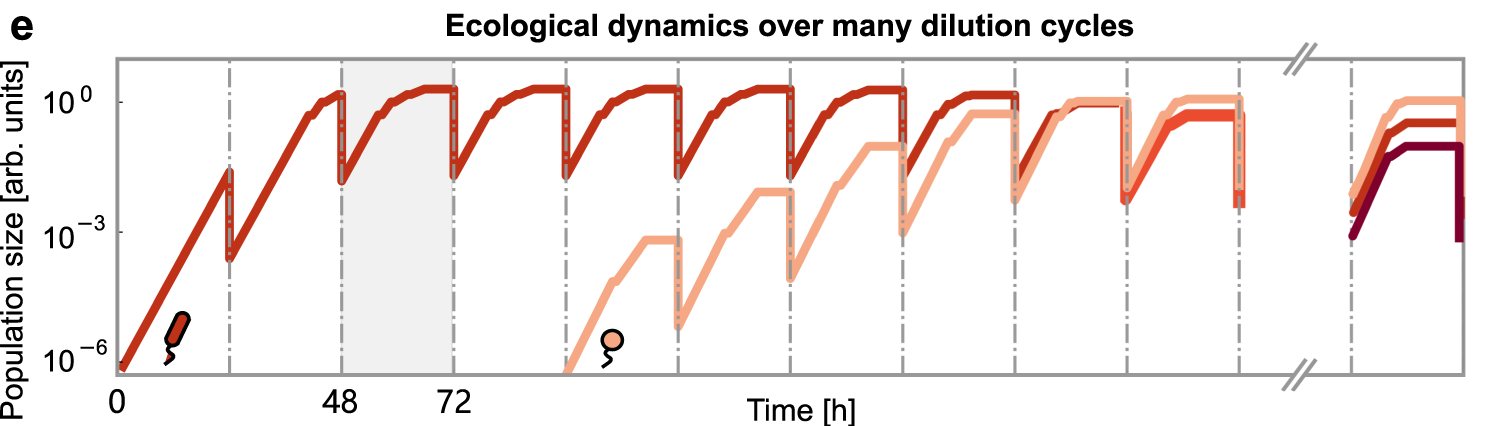
Population dynamics in a serial dilution experiment. During community assembly, new species are added one by one from a species pool. After each successful invasion, the system undergoes several growth-dilution cycles until it reaches a new steady state. - In the second type of a community (eLife 2019) microbes compete for several types of essential nutrients (e.g., sources of C and N) each represented by multiple metabolites (different sugars, amino-acids, NH4, NO3, NO2, etc.)

Network of 8633 possible regime shifts between pairs of 893 uninvadable dynamically stable states in the model with 6 carbon metabolites, 6 nitrogen metabolites and 36 species - In the third type of a community (mSystems 2019) two microbial species not only compete with each other for the same nutrient sources but are also susceptible to the same phage. We identified conditions when this ecosystem could have two stable states separated by a regime shift.
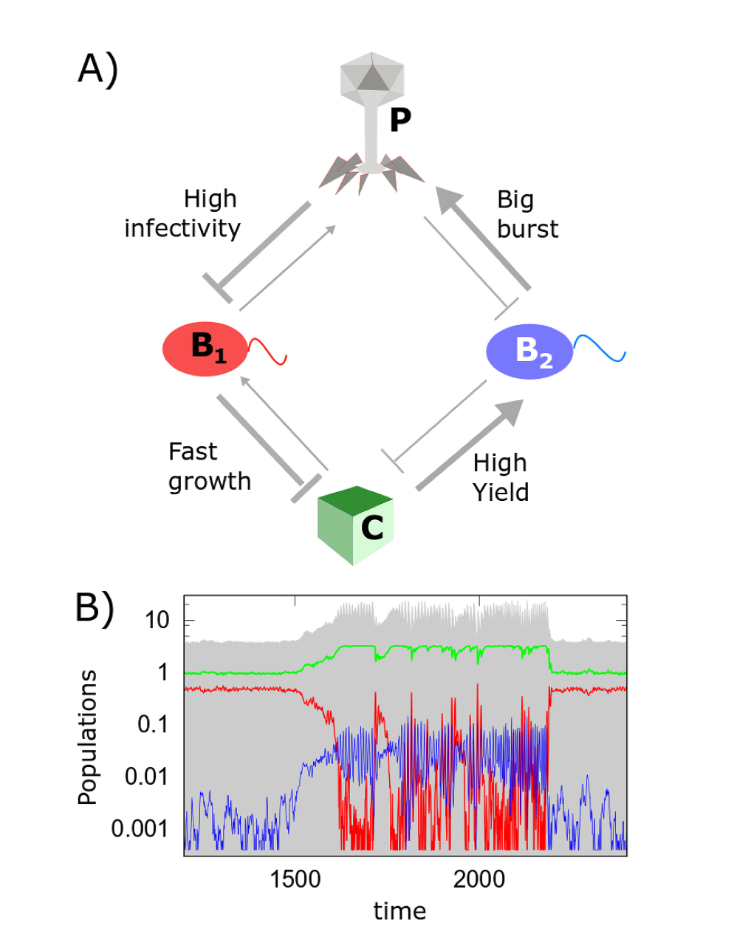
We also studied the population dynamics of phages and their bacterial hosts with the focus on co-evolution of CRISPR immunity (Nat Ecol Evol 2020), sudden population collapses (Sci Rep 2015, PLoS Comp Bio 2015, Sci Rep. 2017) and spatial dynamics of phage epidemics in chemotaxing bacteria (ISME J 2020).
Origin of life
How exactly long functional polymers such as ribozymes emerged out of a primordial soup of nucleotides and oligomers at the dawn of life remains a mystery. In (JCP 2015) we filled a small gap in this puzzle by considering a problem of autocatalysis of polymers capable of template-assisted ligation and driven by cyclic changes in the environment. Our central result is the existence of a first order transition between the regime dominated by short oligomers and another one dominated by long polymers. Another key result is the emergence of the optimal sequence overlap length between a template and its two substrates. The length of this overlap is defined by hybridization kinetics.
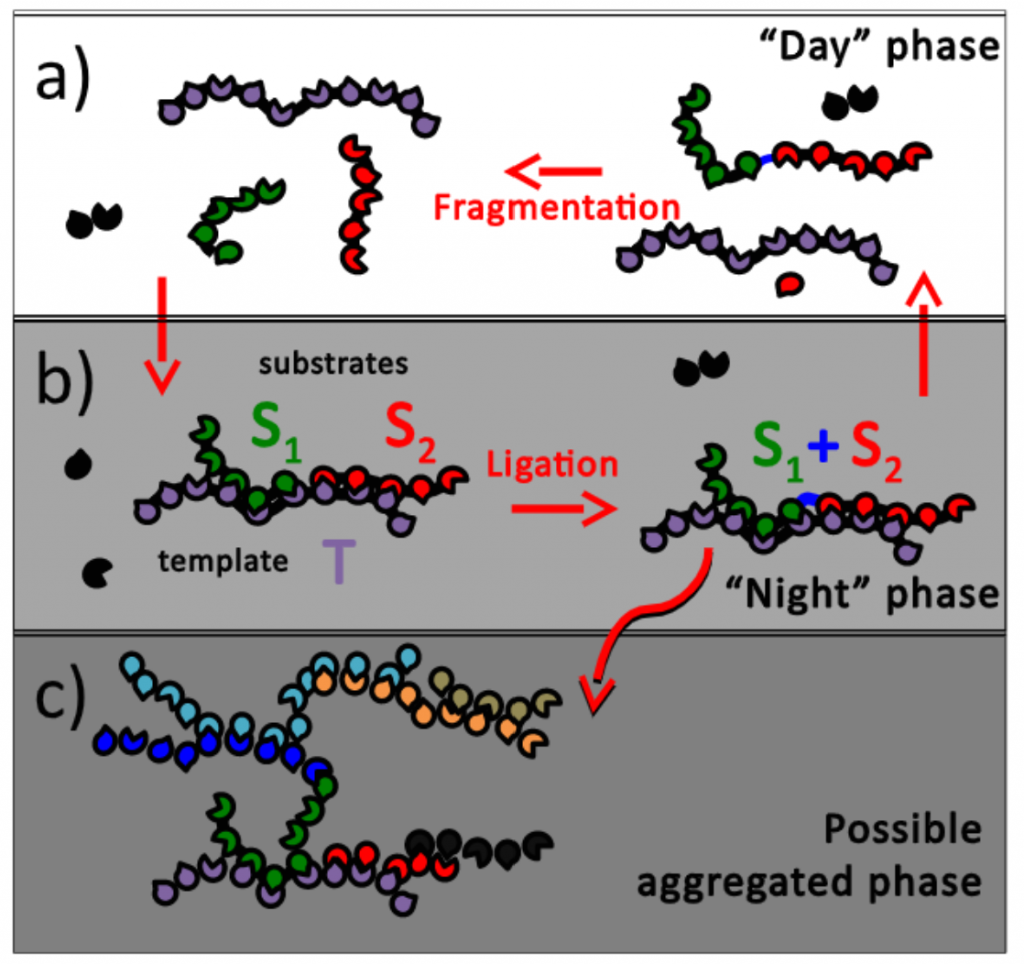
This work was continued in (JCP 2017) where we demonstrated that due to the competition for monomers, only a small subset of polymer sequences survives. More precisely, no matter how large is Z (the monomer alphabet size), only 2L out of ZL possible polymer sequences of length L would ultimately survive. Mathematically, this rule for polymer survival is very similar to the competitive exclusion principle in microbial ecosystems, according to which the number of surviving species in a steady state of an ecosystem cannot be larger than the number of nutrients they feed on. 2-mers (sequences of two letters within a polymer) correspond to microbial species, while 2Z letters on right (Z letters) and left (another Z letters) ends of polymers – to nutrients they use. This parallel ultimately allowed us to introduce and solve a model (eLife 2019) of microbial ecosystems with species competing for two types of essential nutrients (e.g., C and N) corresponding to left and right ends of polymer chains.
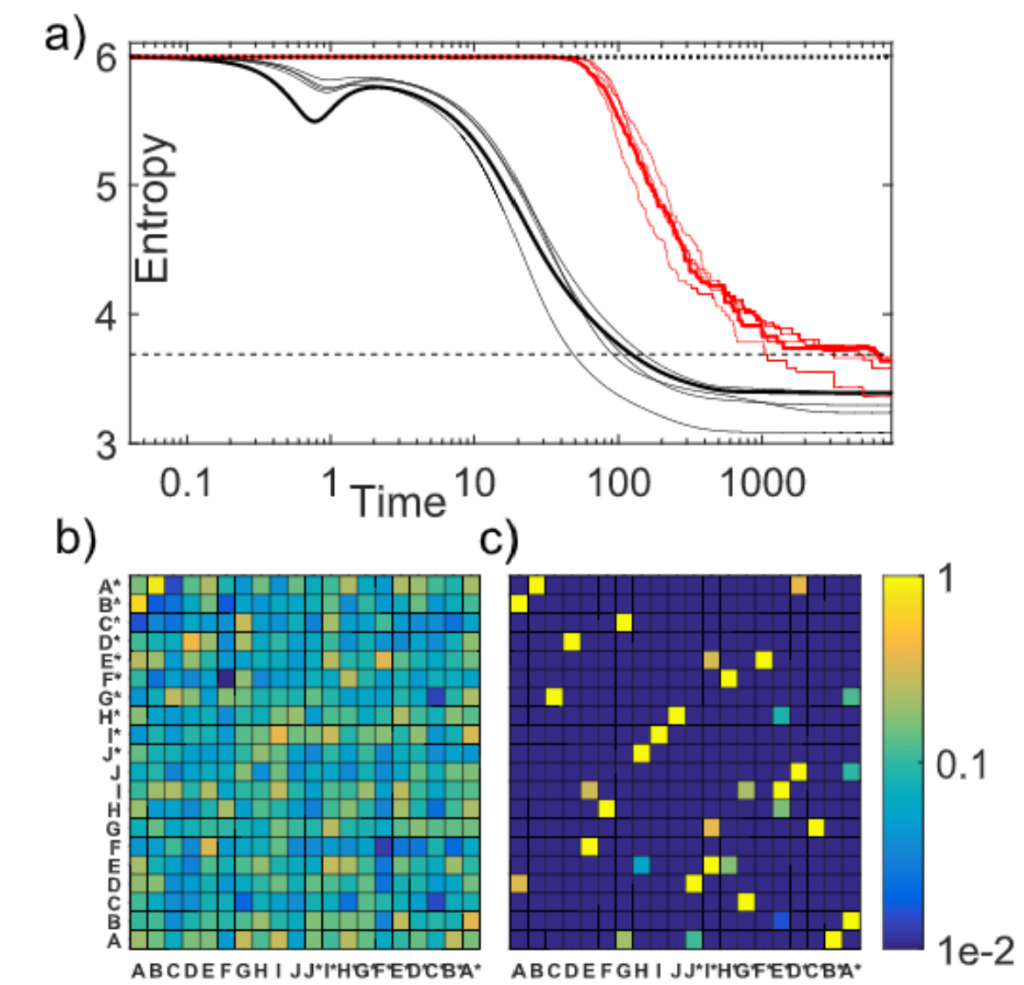
We then applied our theory to explain the results of a recent expriment in Dieter Braun’s lab at Munich (PNAS 2021 Feb). They were assembling longer polymers out of DNA 12-mers made our of letters A and T. The alphabet size in this case is Z=2^12=4096. The main difference with our theory is that selection did not have enough time to eliminate losing sequences. However, the signature of a complex selection landscape driven (among other things) by avoidance of hairpins is already clearly visible. We found the robust creation of long, highly structured sequences with low entropy. At the ligation site, complementary and alternating sequence patterns have developed. However, between the ligation sites, we found either an A-rich or a T-rich sequence within a single oligonucleotide. What emerged was a network of complementary sequences that acted both as templates and substrates of the reaction.
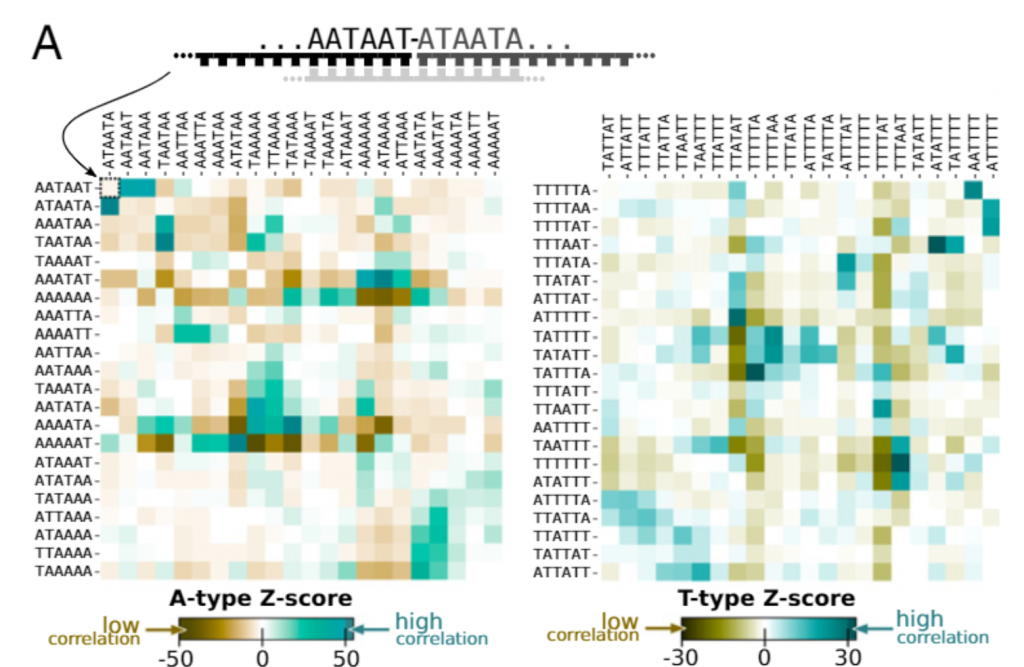
The findings showed that replication by random templated ligation from a random sequence input will lead to a highly structured, long, and nonrandom sequence pool. This is a favorable starting point for a subsequent Darwinian evolution searching for higher catalytic functions in an RNA world scenario.
Deep learning
The question of how deep learning networks acquire the ability to make successful prediction on test data without overfitting training data fascinates me. I am also intrigued by the phenomenon of adversarial examples in which imperceptibly small changes, e.g., to an image of a cat can make deep learning algorithm to confidently classify it as, e.g., guacamole. My group and I are slowly building statistical physics -based understanding of these phenomena.
In the meantime, I participated in a few practical applications of machine learning and was involved in knowledgebase development. In (Xia et al. 2022, Xia et al. 2018) and we compared the performance of multiple deep learning algorithms in predicting tumor cell line response to drug pairs. In (Nambiar et al. 2020) we developed Protein RoBERTa – a transformer neural network algorithm fine-tuned to solve two different protein prediction tasks: protein family classification and protein interaction prediction. In (Arkin et al. 2018) we designed the v1.0 of the DOE Systems Biology Knowledgebase (KBase), an open-source software and data platform that enables data sharing, integration, and analysis of microbes, plants, and their communities.
Epidemiology
During the first wave of COVID-19 pandemic I shifted the focus of my research towards epidemiology. I was one of two founding members of the COVID-19 modeling team for the office of the Governor of Illinois. Between April 2020 and July 2021, our team provided weekly modeling predictions such as the likelihood to exceed hospital capacity in each of the 11 regions of the state.
Parallel to this our team developed an agent-based model of COVID-19 epidemics on the UIUC campus. The model was used to advise the university leadership on testing frequency, scheduling, and mitigation measures such as, e.g., a temporary ban on student parties. For this work I was awarded a Presidential Award and Medallion of the University of Illinois and became the butt of the joke in an XKCD cartoon.
On the scientific front in (PNAS 2021 Apr, eLife 2021) my collaborators and I developed a new type of epidemiological model with stochastic social activity and applied it to describe successive waves of COVID-19 epidemic in US during Summer 2020-Winter 2021. Mark Lipsitch, who was the editor of (eLife 2021) in his evaluation of this work printed alongside the article stated: “This is an excellent and elegant example of what theory can do at its best in epidemiology. … This should stimulate much further work in the field.”. In (PRX 2020) we calibrated the age-of-infection model to multiple data streams (cases, hospitalizations, deaths) to describe the COVID-19 epidemic dynamics in Illinois.
Cuiriously, my first exprience of working on epidemiology predactes COVID-19. In (PRE 2017) we considered the aftermath of a spillover of a pathogen from between two species such as, e.g., bats and humans.
Evolution of bacterial genomes
In (JMB 2009, PNAS 2015) we developed scalable computational algorithms to estimate core and pan-genomes of bacterial species and to analyze Single-Nucleotide Polymorphisms (SNP) within this core genome. Our algorithm separates vertically inherited, clonal, segments from recombined (horizontally transferred) ones. For closely related pairs of E. coli strains, we identified a patchwork of long recombined segments interspersed among clonally inherited genomic segments. Once sequence divergence between strains exceeds ~1.3%, clonal segments virtually disappear. Our results implicate generalized transducing phages in horizontal transfer of genomic segments between strains. In (Genetics 2017) we modeled the evolutionary dynamics based on these observations and applied the model to define stability and boundaries of 12 bacterial species. Biomedical applications of our findings include understanding the emergence and spread of pathogenic bacterial strains (e.g., E. coli) and of antibiotic resistance in bacterial populations.
In (PNAS 2009) we proposed the “toolbox” model of co-evolution of metabolic and regulatory networks by Horizontal Gene Transfer in bacterial and archaeal genomes. Our model explained a number of trends in properties of these networks with genome size. These insights into modular properties of bacterial genomes and networks are important for bioengineering and biomedical applications. In (PLoS Com Bio 2011) we extended this toolbox model to include anabolic (biosynthetic) pathways. To this end we came up with a computational algorithm predicting the minimal biosynthetic pathway to add to the existing metabolic network of an organism so that it can synthesize a desired target metabolite. Algorithms proposed in this paper are relevant for synthetic biology applications. In (NAR 2017; NAR 2011; PNAS 2013) we identified functional and evolutionary determinants of sizes of gene families and the frequency with which they are encoded in bacterial genomes.
The full list of my publications: